Прошло уже почти 20 лет с тех пор как были открыты вирусы иммунодефицита человека типа 1 в 1983 г.
(1, 2) и типа 2 в 1986 г. (3) и установлено, что они являются первопричиной синдрома приобретенного
иммунодефицита (СПИДа). Во всем мире большинство случаев СПИДа сегодня вызвано ВИЧ-1, поэтому
говоря о ВИЧ мы будем подразумевать именно вирус первого типа. Число инфицированных ВИЧ-1 на
земном шаре превышает 40 млн человек, большинство из них живет в Азии, Западной, Экваториальной и
Южной Африке и Южной Америке.
С введением в схемы антиретровирусной терапии в 1995 г. ингибиторов протеазы и ненуклеозидных ингибиторов обратной транскриптазы началась эра высокоактивной антиретровирусной терапии (ВААРТ).
Она ознаменовалась значительным снижением смертности и числа осложнений у ВИЧ-инфицированных,
включая оппортунистические инфекции и злокачественные новообразования. Однако несмотря на все
успехи антиретровирусной терапии последних десяти лет, искоренение вируса у ВИЧ-инфицированных
по-прежнему остается недостижимым.
Лечение ВИЧ-инфекции столкнулось и с новыми проблемами — ранней и отдаленной токсичностью
препаратов и устойчивостью вируса как у отдельных больных, так и в целом. В большинстве стран
Юго-Восточной Азии и Африки заболеваемость и распространенность ВИЧ-инфекции выше, чем в Европе и Северной Америке и продолжают расти. Из-за высокой стоимости лечения и слаборазвитой сети
здравоохранения в развивающихся странах применение ВААРТ пока ограничено. Дальнейший ход пандемии ВИЧ-инфекции во многом зависит от того, смогут ли развивающиеся страны с высокой распространенностью ВИЧ-инфекции в полной мере воспользоваться достижениями Европы и Северной Америки и от того, появится ли в ближайшем будущем вакцина для профилактики этой инфекции.
Главное условие, без которого невозможно ни упрочить завоеванных позиций в лечении ВИЧ-инфекции,
ни достичь новых — будь то иммунотерапия или получение вакцин, — это понимание иммунопатогенеза
ВИЧ-инфекции. Не вызывает сомнений, что течение ВИЧ-инфекции зависит от свойств как возбудителя,
так и макроорганизма.
Течение ВИЧ-инфекции отличается крайним непостоянством — у одних людей инфекция прогрессирует
быстро, у других медленно — даже при заражении от одного и того же источника (4). У некоторых ВИЧ-
инфицированных число лимфоцитов CD4 не снижается и СПИД не развивается в течение 7 и более
лет — в таких случаях (их около 5%) говорят о длительном непрогрессирующем течении инфекции. В
некоторых из этих случаев обнаружены дефектные вирусы с ослабленной способностью к репликации
(5). Однако у большинства ВИЧ-инфицированных вирус активно реплицируется, а различия в скорости
развития иммунодефицита объясняются особенностями макроорганизма.
С выяснением этих особенностей, в том числе генетических факторов и механизмов иммунной защиты,
связывают перспективы дальнейшего изучения патогенеза ВИЧ-инфекции и создания методов иммунотерапии и профилактики.
ВИЧ относится к семейству ретровирусов, подсемейству лентивирусов. Лентивирусы вызывают хронические инфекции с длинным латентным периодом, персистирующей репродукцией вируса и поражением
ЦНС. Возбудители типичных лентивирусных инфекций — вирус висны, вызывающий заболевание у
овец, вирус иммунодефицита обезьян и вирус кошачьего иммунодефицита.
С помощью электронной микроскопии показано, что ВИЧ-1 и ВИЧ-2 имеют сходную структуру. В то же
время у них есть и отличия — по молекулярной массе белков и некоторым дополнительным генам. Филогенетически ВИЧ-2 ближе к вирусу иммунодефицита обезьян, обнаруженному у воротничковых мангобеев, чем к ВИЧ-1. Предполагают, что у людей инфекция, вызванная ВИЧ-2, появилась в результате
заражения от обезьян. Репликация как ВИЧ-1, так и ВИЧ-2 происходит в лимфоцитах CD4; оба вируса
вызывают СПИД, хотя у инфицированных ВИЧ-2 он обычно протекает легче.
Диаметр ВИЧ-1 составляет 100 нм. Снаружи вирус окружен липидной мембраной, в которую встроены 72 гликопротеидных комплекса. Каждый из этих комплексов образован тремя молекулами поверхностного гликопротеида (gp120) и тремя трансмембранного (gp41). Связь между gp120 и gp41 довольно слабая, и поверхностный гликопротеид может спонтанно отсоединяться от вируса. Поэтому gp120 обнаруживается в сыворотке (6), а также лимфоидной ткани ВИЧ-инфицированных (7). При отпочковывании ВИЧ от клетки ее мембранные белки, в том числе HLA классов I и II, и молекулы адгезии, в частности ICAM-1, встраиваются в липидную мембрану вируса. Эти белки облегчают адгезию вируса к клеткам-мишеням. Внутри к липопротеидной оболочке прилежит матриксный белок p17. Сердцевину вируса (капсид) составляет капсидный белок p24, который окружает белковонуклеиновый комплекс: две молекулы вирусной РНК, связанные с протеидом p7 и обратной транскриптазой p66. Вирус содержит все необходимые ферменты для репликации: обратную транскриптазу, интегразу p32 и протеазу p11 (рис. 1) (подробнее см. 8).
Репродукция большинства ретровирусов определяется тремя генами: gag, pol и env (рис. 2). Название генов произошло от кодируемых ими белков: gag — "group-antigen" (капсидный белок), pol — "polymerase"
(полимераза), env — "envelope" (белки внешней оболочки) (подробнее см. 9). Классическая схема генома
ретровирусов записывается как 5'LTR-gag-pol-env-LTR 3'. Оба конца генома содержат длинные концевые
повторы LTR (от англ. long terminal repeat), которые обеспечивают интеграцию в клеточный геном и вирусных белков не кодируют. Гены gag и env кодируют белки капсида и внешней оболочки, ген pol — обратную транскриптазу и другие ферменты. Геном ВИЧ-1 в составе своих 9000 пар нуклеотидов содержит
еще шесть генов — vif, vpu, vpr, tat, rev и nef. Ранее гены nef, vif, vpr и vpu называли дополнительными,
поскольку репликация in vitro возможна и без их участия. В последние годы функция этих генов и кодируемых ими белков стала более понятна. Установлено, что продукты генов nef, tat и rev синтезируются в
ранней фазе репликации ВИЧ.
Регуляторные белки Tat и Rev накапливаются в ядре и связываются с определенными участками вирусной РНК: первый с трансактивируемым регуляторным элементом (TAR) в области длинных концевых
повторов, второй — с Rev-чувствительным регуляторным элементом (RRE) в области гена env. Белок Tat
активирует транскрипцию промоторной области длинных концевых повторов и необходим для репликации вируса почти во всех культурах клеток. Белок Tat нуждается в клеточном кофакторе — циклине T1
(10). Белки Tat и Rev стимулируют транскрипцию провирусной ДНК в РНК, элонгацию РНК и транспорт
РНК из ядра в цитоплазму и необходимы для трансляции. Белок Rev обеспечивает также транспорт компонентов вируса из ядра и переключение синтеза регуляторных белков на синтез структурных.
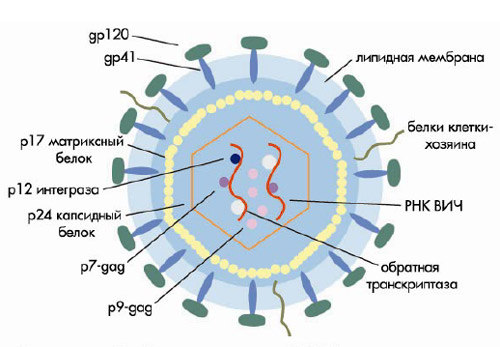
Рисунок 1.Схема строения ВИЧ (пояснения в тексте)
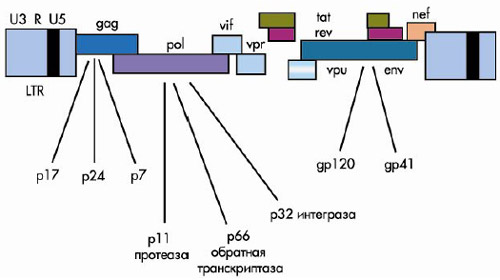
Рисунок 2. Геном ВИЧ (пояснения в тексте)
Белок Nef выполняет несколько функций. Он подавляет экспрессию молекул CD4 (11) и HLA классов I и
II (12) на поверхности инфицированных клеток, и тем самым позволяет вирусу ускользать от атаки цитотоксических T-лимфоцитов CD8 и от распознавания лимфоцитами CD4. Белок Nef может также угнетать
активацию T-лимфоцитов, связываясь с различными белками — компонентами систем внутриклеточной
передачи сигнала (подробнее см. 13).
У инфицированных вирусом иммунодефицита обезьян макак-резус активная репликация вируса и прогрессирование болезни возможны только при интактном гене nef. Делеции гена nef были обнаружены в
штаммах ВИЧ, выделенных у группы австралийцев с длительным непрогрессирующим течением инфекции (5). Однако у части из них со временем появились признаки прогрессирования инфекции, в том числе снижение числа лимфоцитов CD4. Таким образом, хотя делеции гена nef и могут замедлять репликацию вируса, гарантией от СПИДа они не служат.
Белок Vpr необходим для репликации вируса в непролиферирующих клетках, в том числе макрофагах.
Этот белок наряду с другими клеточными и вирусными промоторами активирует длинные концевые повторы генома ВИЧ. Недавно выяснено, что белок Vpr играет важную роль в переносе провируса в ядро
(подробнее см. 14) и вызывает задержку пролиферации клетки в периоде G2.
Белок Vpu важен для отпочковывания вируса из клетки: мутации гена vpu приводят к накоплению вирусных частиц у внутренней поверхности клеточной мембраны. Этот белок участвует также в разрушении комплексов CD4-gp160 в эндоплазматическом ретикулуме, позволяя тем самым gp160 включаться в
формирование новых вирионов (15).
Согласно последним публикациям белок Vif играет важную роль в поддержке репликации вируса (16).
Штаммы, лишенные этого белка, не реплицируются в лимфоцитах CD4, некоторых линиях
T-лимфоцитов («недоступных клетках») и макрофагах. Эти штаммы способны проникать в клетки-мишени и начинать обратную транскрипцию, однако синтез провирусной ДНК остается незавершенным. In vitro слияние «доступных» и «недоступных» клеток приводит к «недоступному» фенотипу; это
означает, что репликация ВИЧ зависит от наличия или отсутствия клеточного ингибитора. Недавно такой эндогенный ингибитор был выявлен — APOBEC3G (17). APOBEC3G (от англ. «apolipoprotein B
mRNA editing enzyme catalytic polypeptide like 3G» — фермент-каталитический полипептид, корректирующий мРНК апопротеина B, типа 3G») принадлежит к семейству внутриклеточных ферментов, специфически дезаминирующих цитозин в урацил в составе мРНК или ДНК, что приводит к накоплению
мутаций G/A и распаду вирусной ДНК. Образуя комплекс с APOBEC3G, Vif блокирует ингибиторную
активность APOBEC3G (рис. 3a). Примечательно, что противовирусная активность APOBEC3G очень
специфична для разных видов животных, а блокада APOBEC3G белком Vif очень специфична для ВИЧ.
Образуемый ВИЧ-1 белок Vif не образует комплекса с APOBEC3G мышей и макак-резус. В отсутствие
Vif APOBEC3G встраивается в образующиеся вирусные частицы и затем проникает в клетки-мишени,
блокируя провирусную ДНК (рис. 3б). При наличии же белка Vif APOBEC3G связывается, распадается и
не встраивается в новые вирусные частицы. APOBEC3G синтезируется в лимфоцитах и макрофагах —
основных мишенях ВИЧ-инфекции.
В настоящее время остается много вопросов о регулировании внутриклеточного APOBEC3G. Например,
есть ли критическое количество внутриклеточного APOBEC3G, ограничивающее ВИЧ-инфекцию в присутствии Vif, и существует ли генетический полиморфизм APOBEC3G, способный влиять на течение болезни. В то же время уже описаны эпитопы, с помощью которых происходит взаимодействие Vif и
APOBEC3G, и пути внутриклеточного распада комплекса APOBEC3G-Vif. Следует заметить, что специфические ингибиторы, блокирующие взаимодействие Vif и APOBEC3G или препятствующие внутриклеточному распаду APOBEC3G, в будущем могут стать перспективным методом лечения. В принципе риск
развития устойчивости при блокаде клеточных структур минимальный, поэтому, воздействие на Vif и
APOBEC3G представляется привлекательным методом лечения.
В целом эти данные объясняют не только почему Vif необходим для репликации ВИЧ, но почему репликация ВИЧ видоспецифична. Помимо APOBEC3G был открыт и другой клеточный фактор (см. ниже),
который тоже может объяснять видоспецифичность репликации вируса.
Ключевая роль APOBEC3G и других цитидиновых дезаминаз может не ограничиваться ВИЧ-1. Накопление мутаций G/A было обнаружено у различных штаммов вируса гепатита B. In vitro накопление ДНК
вируса гепатита B резко снижалось в присутствии APOBEC3G, но при трансфекции vif этот эффект был
обратим (18).
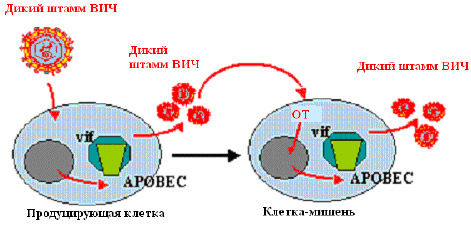
Рис.3а
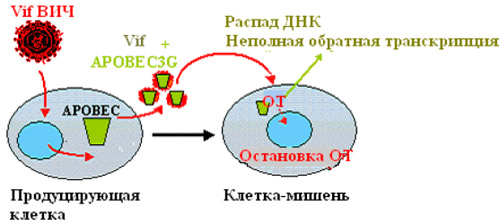
Рис. 3б
Рисунок 3. Инфекция, вызванная диким штаммом ВИЧ: Vif взаимодействует с APOBEC3G, связывается с ним и предотвращает его встраивание в новые вирусы (рис. 3а). Штаммы ВИЧ с делецией vif не способны подавить внутриклеточный APOBEC3G, который затем встраивается в новые вирусы и обрывает их обратную транскрипцию в клетке-мишени. ОТ — обратная транскрипция.
CD4 — главный рецептор ВИЧ
CD4 — это мономерный гликопротеид массой 58 кДа, который обнаруживается на поверхности примерно 60% T-лимфоцитов, предшественников T-лимфоцитов в костном мозге и тимусе, а также моноцитов,
макрофагов, эозинофилов, дендритных клеток и клеток микроглии ЦНС. Внеклеточный участок CD4 на
T-лимфоцитах состоит из 370 аминокислот, гидрофобный трансмембранный и внутриклеточный участки — из 25 и 38 аминокислот соответственно. Внеклеточный участок содержит четыре иммуноглобулиноподобных домена D1-D4 и область V2 (аминокислоты 40-55), играющую важную роль в присоединении gp120 к CD4. Эта область перекрывается с участком связывания молекул HLA класса II, которые являются естественными лигандами CD4.
Идентификация на лимфоцитах CD4 участка связывания gp120 подтолкнула к попыткам использования
свободных молекул CD4 (sCD4) с целью нейтрализации циркулирующего вируса и предупреждения за-
ражения новых клеток. Однако эти попытки провалились: хотя лабораторные штаммы легко нейтрализовались sCD4, нейтрализации штаммов, полученных от больных, добиться не удалось. Более того, оказалось, что sCD4 способны вызывать конформационные изменения внешней оболочки вируса, которые облегчали заражение клеток-мишеней (19).
Молекулы CD4 связываются с антигенраспознающими рецепторами T-лимфоцитов и молекулами HLA
класса II на поверхности антигенпредставляющих клеток. Связывание gp120 с CD4 не только служит
ключевым моментом в проникновении вируса в клетку, но и нарушает внутриклеточную передачу сигнала и вызывает апопотоз лимфоцитов CD4 (20). В последние два года возобновился интерес к идее блокирования CD4 как основного клеточного рецептора ВИЧ. PRO542 представляет собой полученный генно-инженерными методами тетравалентный белок слияния CD4-IgG2, который не только подавлял репликацию in vitro, но и показал впечатляющую противовирусную активность у больных с высокой вирусной нагрузкой в первых клинических испытаниях (21).
О том, что молекула CD4 является главным и необходимым рецептором для ВИЧ-1, ВИЧ-2 и вируса иммунодефицита обезьян, было известно уже в 1984 г. (22, 23). Однако позже с помощью трансфекции человеческих CD4 в клеточные линии животных было показано, что одних только молекул CD4 на поверхности клеток для проникновения ВИЧ недостаточно. Был сделан вывод о существовании дополнительных рецепторов — корецепторов. Оказалось также, что некоторые лабораторные штаммы ВИЧ-1, а также штаммы ВИЧ-2 и вируса иммунодефицита обезьян могут проникать в клетки, лишенные молекул
CD4. Примечательно, что моноклональные антитела к CD4 индуцируют связывание конформационных
эпитопов (CD4I) с gp120 этих CD4-независимых штаммов. Это позволило предположить, что gp120
CD4-независимых штаммов уже несут области, необходимые для распознавания корецепторов, поэтому
связывание с CD4 не обязательно для проникновения этих штаммов в клетку. CD4-независимые штаммы
легко нейтрализуются сывороткой ВИЧ-инфицированных, следовательно они являются мишенью им-
мунного ответа (22).
Рецепторы хемокинов — корецепторы ВИЧ
Важной вехой в изучении начальной стадии проникновения ВИЧ-1 в клетку стало открытие, сделанное в
1995 г. Cocchi и коллегами: в совместных культурах лимфоциты CD8, полученные от ВИЧ-
инфицированных, способны подавлять репродукцию ВИЧ в аутологичных и аллогенных лимфоцитах
CD4, причем этот эффект не связан с цитотоксичностью (25). В супернатанатах лимфоцитов CD8, взятых
у ВИЧ-инфицированных, были обнаружены хемокины MIP-1?, MIP-1? и RANTES. Было показано, что
эти хемокины могут дозозависимо подавлять репродукцию некоторых штаммов вируса (26). MIP-1?,
MIP-1? и RANTES — это естественные лиганды рецептора хемокинов CCR5. Спустя несколько месяцев
после сообщения Cocchi несколькими исследователями было показано, что CCR5 служит необходимым
корецептором для моноцитотропных (M-тропных) штаммов ВИЧ-1 (27, 28, 29). За несколько недель до
этого было выяснено, что корецептором для T-лимфоцитотропных (T-тропных) штаммов ВИЧ служит
рецептор хемокинов CXCR4 (фузин) (28). Моноцитотропные (M-тропные) штаммы ВИЧ-1 — это штаммы, которые хорошо размножаются в культурах макрофагов, но не способны заражать T-клеточные линии (т.е. перевиваемые культуры T-лимфоцитов), однако легко инфицируют первичные культуры
T-лимфоцитов крови. T-тропные штаммы, наоборот, хорошо культивируются в T-клеточных линиях и
плохо — в макрофагах, и легко инфицируют первичные культуры T-лимфоцитов крови. Таким образом,
следует отметить, что M-тропные и T-тропные штаммы ВИЧ-1 легко заражают первичные культуры
T-лимфоцитов человека.
Хемокины (хемотаксические цитокины) и их рецепторы изначально были описаны как факторы, усиливающие миграцию лейкоцитов (хемотаксис) и их провоспалительную активность. Хемокины представляют собой белки, имеющие в составе 68-120 аминокислот. В зависимости от порядка цистеиновых последовательностей хемокины делят на C-X-C (?-хемокины), C-C (?-хемокины) и C-хемокины. Хемокины
гомологичны между собой по структуре и могут связываться с одними и теми же рецепторами. Хемокины действуют через рецепторы, имеющие семь трансмембранных доменов и сопряженные с G-белками.
Естественный лиганд рецептора хемокинов CXCR4 — фактор SDF-1 (от англ. «stromal cell-derived
factor» — выделенный из стромальных клеток). Этот фактор предотвращает заражение активированных
лимфоцитов CD4 T-тропными штаммами ВИЧ. Хемокины RANTES (от англ. Regulation-upon-Activation,
Normal T Expressed and Secreted — молекулы «регуляции при активации», экспрессируемые и секретируемые нормальными T-лимфоцитами) и макрофагальные воспалительные белки MIP-1? и MIP-1? (от
англ. macrophage inhibitory protein) являются естественными лигандами рецептора CCR5 и способны
предотвращать инфицирование T-лимфоцитов M-тропными штаммами ВИЧ. Роль корецепторов показана на рис. 4: T-тропные штаммы ВИЧ заражают преимущественно активированные лимфоциты CD4 крови и клеточные линии, используя для входа в клетку рецепторы CXCR4 и CD4. M-тропные штаммы способны заражать лимфоциты CD4, моноциты и макрофаги и используют для входа в клетку рецепторы
CCR5 и CD4.
Взаимодействие gp120 и клеточных рецепторов сегодня стали более понятны. Гликопротеид gp120 сначала связывается с определенными эпитопами CD4. После этого gp120 претерпевает конформационные
изменения, обеспечивающие более эффективное взаимодействие его петли V3 с соответствующим корецептором. От связывания gp120 с корецептором зависит слияние внешней оболочки вируса с клеточной
мембраной. Трансмембранный гликопротеид gp41 (часть гликопротеида внешней оболочки вируса
gp160) играет ключевую роль в слиянии внешней оболочки вируса и клеточной мембраны подобно гемагглютинину вируса гриппа. После связывания gp120 с рецептором CD4, в gp41 происходят конформационные изменения, в результате которых гидрофобный N-концевой фрагмент gp41 внедряется в мембрану клетки-мишени.
Предположение о том, что gp41 действует наподобие пружинной защелки, было подтверждено с помощью кристаллографического анализа (31). После того, как были расшифрованы ключевые для этого процесса аминокислотные последовательности, усилия были направлены на синтез пептидов, способных
связываться с доменами gp41, необходимыми для индукции конформационных изменений. Такое связывание должно предотвращать слияние внешней оболочки вируса с клеточной мембраной.
Первым из пептидов, связывающих gp41 и включенных в клинические испытания для оценки подавления репродукции вируса, был T-20 (энфувиртид) (32). В настоящее время энфувиртид уже используется
для лечения некоторых групп больных. Его недостаток — невозможность приема внутрь: препарат вводится в инъекциях.
С помощью трансфекции клеточных линий были открыты и другие рецепторы хемокинов, которые наряду с CCR5 и CXCR4 служат корецепторами для некоторых штаммов ВИЧ — CCR3, CCR2, CCR8, CCR9,
STRL33 («Bonzo»), Gpr 15 («Bob»), Gpr 1, APJ и ChemR23 (33, 34). По некоторым данным, APJ служит
корецептором ВИЧ для проникновения в клетки ЦНС. Несмотря на широкий спектр корецепторов
ВИЧ-1, in vivo основное значение, по-видимому, играют CCR5 и CXCR4.
Важность CCR5 как ведущего корецептора M-тропных штаммов ВИЧ становится видна из следующего
наблюдения: большинство людей с дефектным геном CCR5 не восприимчивы к ВИЧ-1 (35). Эксперименты показали, что лимфоциты этих людей in vitro устойчивы к M-тропным штаммам ВИЧ-1, но заражаются T-тропными штаммами. Лимфоциты этих людей не экспрессируют на своей поверхности рецептор CCR5 вследствие делеции 32 пар нуклеотидов в гене CCR5. Во всем мире известно лишь несколько
случаев ВИЧ-инфекции у гомозигот по делеции гена CCR5. Не удивительно, что все они были вызваны
T-тропными штаммами, корецептором которым служит CXCR4 (36).
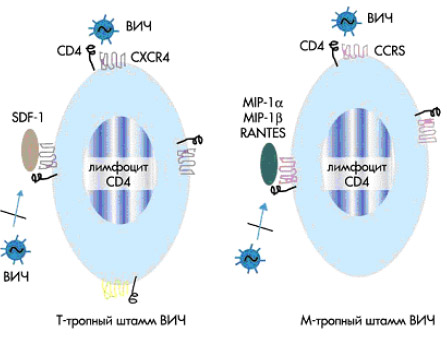
Рисунок 4. Предотвращение проникновения в клетку М-тропных и Т-тропных штаммов ВИЧ путем связывания естественными лигандами рецепторов хемокинов CCR5 и CXСR4.
Эпидемиологические исследования показали, что по делеции гена CCR5 гетерозиготны 10-20% белых
(доля гетерозигот выше у выходцев из северной Европы), а гомозиготны — около 1% белых (37). У чернокожих и азиатов такой делеции гена CCR5 не обнаружено; это говорит о том, что мутация возникла
уже после разделения человечества на расы в процессе эволюции.
У гетерозигот по делеции 32 пар нуклеотидов в гене CCR5 снижена экспрессия рецептора CCR5 на поверхности клеток; показано, что среди них больше лиц с длительным непрогрессирующим течением
ВИЧ-инфекции (37). Кроме того, у ВИЧ-инфицированных гетерозигот по делеции 32 пар нуклеотидов в
гене CCR5 медленнее прогрессирует СПИД, лучше ответ на ВААРТ и меньше частота лимфом. Это говорит о том, что плотность рецепторов CCR5 на поверхности клетки влияет на репликацию ВИЧ не
только in vitro, но и in vivo.
Помимо делеции 32 оснований гена CCR5 были описаны и другие случаи генетического полиморфизма в
отношении рецепторов хемокинов (CCR2) и их промоторов (CCR5), которые влияли на скорость прогрессирования ВИЧ-инфекции (38, 39).
У больных с быстрым прогрессированием инфекции (быстрое падение числа лимфоцитов CD4) штаммы,
использующие в качестве основного корецептора CXCR4, выделяются чаще, чем у больных со стабильным числом лимфоцитов CD4.
Экспрессия корецепторов на лимфоцитах CD4 зависит от уровня активности этих клеток.
Рецептор CXCR4 экспрессируется главным образом на девственных T-лимфоцитах, а рецептор CCR5 —
на активированных T-лимфоцитах, клетках памяти и эффекторных лимфоцитах. В ранней стадии ВИЧ-1-
инфекции обнаруживаются преимущественно M-тропные штаммы ВИЧ. Примечательно, что M-тропные
штаммы ВИЧ передаются чаще независимо от того, какие штаммы — T-тропные или нет — преобладают
у источника инфекции. Пока неясно, обусловлено ли это избирательным транспортом M-тропных штаммов дендритными клетками подслизистого слоя или благоприятствованием местной хемокиновой и цитокиновой среды для репродукции M-тропных вирусов. Привлекают внимание последние исследования
Cheng-Meyer и соавт., выдвинувших гипотезу о том, что благодаря репродукции в макрофагах
M-тропным вирусам легче ускользнуть от иммунной системы, чем T-тропным, что и дает им преимущество в выживании.
В связи с этим блокада рецепторов CCR5 представляется перспективным направлением терапии ВИЧ-
инфекции. Моноклональные антитела к CCR5 (2D7 и другие) in vitro способны блокировать проникновение штаммов, использующих в качестве корецепторов CCR5, в лимфоциты CD4 и макрофаги. Разработаны и уже проходят клинические испытания низкомолекулярные блокаторы рецепторов CCR5. Однако
исследования in vitro, а также эксперименты на мышах линии SCID, показали, что подавление штаммов,
использующих корецепторы CCR5, может приводить к изменению их тропности в сторону корецепторов
CXCR4.
Низкомолекулярные блокаторы, в том числе T-22, ALX40-4C и AMD3100, могут блокировать рецепторы
CXCR4 (40, 41) и в настоящее время проходят доклинические и клинические испытания.
Сегодня разрабатываются методы модулирования экспрессии хемокиновых рецепторов. Интракины —
это хемокины, которые находятся в цитоплазме и могут захватывать и связывать соответствующие рецепторы у поверхности клетки (42). «Короткие интерферирующие РНК» (киРНК) являются новым молекулярным инструментом селективной инактивации генов-мишеней. Двухцепочечная РНК расщепляется
ферментом dicer-1 на короткие части (“21-23-меры”). Эти олигомеры могут комплементарно связываться
с длинными последовательностями РНК, после чего последние распадаются. Такие олигомеры сегодня
обнаружены у растений с противовирусной активностью. Использование киРНК против CCR5 может
предотвратить экспрессию CCR5 in vitro.
Хотя использование блокаторов хемокиновых рецепторов кажется перспективным направлением в лечении ВИЧ-инфекции, немало вопросов еще остаются открытыми. Эксперименты на мышах с инактивированными генами CXCR4 или SDF-1 обнаружили тяжелые нарушения кроветворения и развития мозжечка
(43). Не вызовет ли блокада рецепторов CXCR4 у детей или взрослых нарушений со стороны каких-либо
систем органов, пока не известно.
После слияния вируса с мембраной клетки вирусной ядро высвобождается в цитоплазму. Эти «ранние
события» сегодня подробно изучаются. ВИЧ способен проникать в лимфоциты макак-резус, но до начала
обратной транскрипции или вскоре после него репликация вируса прекращается. Эта внутриклеточная
блокада опосредована клеточным фактором TRIM5?, который является компонентом клеточных органелл. У разных видов животных TRIM5? специфически подавляет определенные ретровирусы. Например TRIM5? макак-резус (TRIM5? rh) сильнее подавляет репликацию ВИЧ, чем TRIM5? человека, в то
время как вирус иммунодефицита обезьян, который в естественных условиях патогенен для макак, менее чувствителен к другим формам TRIM5?. Отчасти это объясняет видовую специфичность ВИЧ к
клеткам человека (44). TRIM5? клеток человека и других приматов может подавлять репликацию других лентивирусов и является новым фактором клеточной устойчивости, биологическое значение которого до конца не ясно. В точности не известно также, как TRIM5? блокирует обратную транскрипцию;
предполагается, что TRIM5? воздействует на капсидный белок проникающего в клетку вируса, вызывая
его полную протеолитическую деградацию.
ВИЧ проникает как в активированные T-лимфоциты, так и в покоящиеся, однако в покоящихся клетках
не завершается синтез вирусной ДНК (45). Синтез провирусной ДНК на матрице вирусной РНК в цитоплазме клетки под действием фермента обратной транскриптазы — это ключевой момент в репродукции
ВИЧ. Блокирование обратной транскриптазы нуклеозидным ингибитором зидовудином было первой попыткой подавить репродукцию вируса у ВИЧ-инфицированных. Начиная с середины 80-х годов арсенал
ингибиторов обратной транскриптазы значительно расширился, и сегодня в клинической практике применяются различные нуклеозидные, нуклеотидные и ненуклеозидные ингибиторы обратной транскриптазы.
Обратная транскрипция происходит в несколько этапов. После связывания праймеров тРНК в участке
PBS (участок связывания праймера) начинается синтез минус-цепи провирусной ДНК, который завершается на 5'-концевом участке с образованием короткой цепи ДНК R/U5. Следующий этап — расщепление
РНК над участком PBS вирусным ферментом РНКазой H и сдвиг рамки ДНК R/U5 с гибридизацией в
области R-участка на 3'-концевом участке РНК. После этого завершаются синтез полноразмерной провирусной ДНК и расщепление тРНК. В результате обратной транскрипции образуется двойная цепь ДНК
ВИЧ с длинными концевыми повторами LTR на каждом конце.
В покоящихся T-лимфоцитах образованная в результате обратной транскрипции провирусная ДНК не
встраивается в геном клетки хозяина. Для того, чтобы клеточная ДНК встроилась в ДНК клетки-хозяина
необходима активация клетки и перемещение вирусного преинтеграционного комплекса из цитоплазмы
в ядро. In vitro активация клеток происходит, например, после стимуляции антигенами или митогенами,
in vivo активация иммунной системы наблюдается после контакта с антигеном, вакцинации или на фоне
оппортунистической инфекции. Кроме того, имеется все больше указаний, что вирусный гликопротеид
gp120 сам способен активировать зараженные клетки, способствуя тем самым встраиванию вирусной
ДНК в клеточный геном. Помимо моноцитов, макрофагов и клеток микроглии невстроенная в клеточный
геном провирусная ДНК ВИЧ содержится в покоящихся лимфоцитах CD4 — долгоживущих клетках, которые являются важным резервуаром ВИЧ и латентной инфекции (46). Поскольку естественное течение
ВИЧ инфекции характеризуется постоянной репродукцией вируса в активированных лимфоцитах CD4,
пребывание вируса в латентном состоянии в покоящихся лимфоцитах CD4 скорее всего является случайным феноменом, не имеющим большого значения в патогенезе этой инфекции. Однако этот небольшой резервуар латентного провируса приобретает особое значение с началом ВААРТ: антивирусные
препараты не действуют на нереплицирующиеся провирусы, поэтому ВИЧ продолжает персистировать в
этих клетках и способен к репродукции и новому витку инфекции при отмене препаратов. Таким образом, существование этого резервуара латентного вируса не позволяет добиться искоренения вируса у
ВИЧ-инфицированных с помощью ВААРТ.
Лишь недавно стало ясно, почему ВИЧ плохо реплицируется в покоящихся лимфоцитах CD4. Клеточный
белок Murr1, участвующий в метаболизме меди, способен подавлять репликацию ВИЧ-1 в нестимулированных лимфоцитах CD4. Murr1 был обнаружен в первичных покоящихся лимфоцитах CD4, в которых
он подавлял активацию фактора транскрипции NF?B, блокируя деградацию I?B?. I?B? предотвращает
миграцию NF?B в ядро, особенно после цитокиновой стимуляции (например, ФНО?). Поскольку области длинных концевых повторов ВИЧ имеют множество сайтов для NF?B, предотвращение миграции
NF?B в ядро должно подавлять репликацию ВИЧ. Ингибирование Murr-1 с помощью киРНК приводит к
репликации ВИЧ в покоящихся лимфоцитах CD4 (47). Персистирование ВИЧ в покоящихся лимфоцитах
CD4 и других клетках-резервуарах считается основной причиной, по которой не удается добиться искоренения вируса. Если оно в принципе достижимо, подробное изучение того, как и когда появляются клеточные резервуары ВИЧ и как на них можно воздействовать, играет первостепенное значение в создании
методов, нацеленных на искоренение инфекции.
Клеточные факторы транскрипции, в частности NF-kB, тоже могут связываться с длинными концевыми
повторами LTR. После стимуляции митогенами или цитокинами фактор NF-kB транслоцируется в ядро,
где связывается с длинным концевым повтором и запускает транскрипцию вирусных генов. В результате
транскрипции сначала синтезируются регуляторные вирусные белки, в частности Tat и Rev. В ядре клетки белок Tat связывается с трансактивируемым регуляторным элементом (TAR), расположенным в начале вирусной РНК, и стимулирует транскрипцию и образование длинных транскриптов РНК. Белок Rev
стимулирует экспрессию генов, отвечающих за синтез структурных белков и ферментов, и подавляет
продукцию регуляторных белков, тем самым запуская образование зрелых вирусных частиц. Белки, кодируемые генами pol и gag, образуют вирусный капсид; продукты гена env образуют поверхностный
гликопротеид gp120 — «шипы» внешней оболочки вируса. Гликопротеид gp120 образуется в результате
расщепления вирусной протеазой своего предшественника — гликопротеида gp160 — на gp120 и gp41.
Продукты гена gag также образуются в результате расщепления протеазой ВИЧ предшественника — полипротеина массой 53 кДа — на белки p24, p17, p9 и p7. Поскольку расщепление молекул-предшественников протеазой ВИЧ — необходимое условие для образование новых вирусных частиц,
этот фермент служит еще одной мишенью для антиретровирусной терапии (48). Сборка вирусов происходит поэтапно: из вирусной РНК, белков Gag и ферментов Pol образуется нуклеокапсид, который пере-
мещается к клеточной мембране. Крупные молекулы-предшественники расщепляются вирусной протеа-
зой, после чего завершается сборка зрелых вирусов и они отпочковываются от клетки. При отпочковы-
вании в липидную оболочку вируса могут встраиваться различные белки клетки-хозяина, фосфолипиды
и холестерин. В отличие от T-лимфоцитов, в которых отпочковывание происходит на поверхности кле-
ток и приводит к выделению вирусов в межклеточное пространство, в моноцитах и макрофагах процесс
завершается накоплением вируса внутри клеточных вакуолей.
Репликация ретровирусов подвержена ошибкам и характеризуется высокой частотой спонтанных мутаций. В среднем при обратной транскрипции происходит от 1 до 10 ошибок на один геном или один цикл
репликации. Мутации могут приводить утрате вирусом способности к репликации. С другой стороны,
могут появляться и накапливаться мутации, в результате которых вирус приобретает устойчивость к антивирусным препаратам. Под давлением противовирусных средств и при неполном подавлении репликации вируса устойчивые вирусы начинают преобладать.
Кроме того, для ВИЧ характерна высокая скорость репликации и, соответственно, большой оборот вирусных частиц: в среднем за сутки образуется и разрушается 1 млрд вирусных частиц. Из-за высокой
скорости репликации вируса и большой частоты мутаций у одного и того же больного накапливается
множество близких вариантов вируса, называемых псевдовидами. В результате естественного отбора
преимущественно сохраняются псевдовиды, приобретшие в результате мутаций устойчивость к антиретровирусным препаратам и факторам иммунной защиты, таким как нейтрализующие антитела и цитотоксические T-лимфоциты.
Дендритные клетки как основные антигенпредставляющие клетки
Дендритные клетки, макрофаги и B-лимфоциты — это основные антигенпредставляющие клетки иммунной системы. Дендритные клетки служат наиболее сильными индукторами специфического иммунного ответа и являются необходимым элементом для начала первичных антиген-специфических иммунных реакций. Предшественники дендритных клеток мигрируют из костного мозга в периферические органы иммунной системы и подслизистый слой кишечника, мочеполовой и дыхательной систем. Они способны захватывать и перерабатывать растворимые антигены и мигрировать во вторичные органы иммунной системы, где активируют антиген-специфические T-лимфоциты.
Дендритные клетки — это гетерогенная группа клеток с различными функциональными возможностями
и фенотипическими маркерами в зависимости от микроокружения и степени зрелости. Незрелые дендритные клетки могут захватывать и перерабатывать антигены, но их способность активировать
T-лимфоциты слаба. Зрелые дендритные клетки, напротив, обладают в основном иммуностимулирующей активностью. Тканевые дендритные клетки и клетки Лангерганса (специализированные клетки кожи
и слизистых) имеют незрелый фенотип и могут захватывать антиген. После этого они мигрируют в лимфоидную ткань, где приобретают зрелый фенотип.
Для стимуляции лимфоцитов CD8 и образования антиген-специфических цитотоксических
T-лимфоцитов необходимо представление пептидного антигена в комплексе с HLA класса I. Дендритные
клетки могут заражаться вирусами, например вирусом гриппа. При этом в цитоплазме клетки синтезируются вирусные белки, которые расщепляются на пептиды и переносятся в цитозоль эндоплазматического ретикулума, где связываются с HLA класса I. Образовавшийся комплекс перемещается на поверхность дендритной клетки. Число комплексов специфический пептидный антиген — HLA класса I обычно
ограничено, и поэтому антиген распознается лишь одним клоном T-лимфоцитов из 100 000 и более. Антигенраспознающие рецепторы T-лимфоцитов (TCR) обладают низкой аффинностью к антигену (1 mM и
менее). Высокая плотность дополнительных стимулирующих молекул на поверхности дендритных клеток позволяет усилить взаимодействие антигенраспознающих рецепторов с комплексом пептидный антиген — HLA и тем самым запустить пролиферацию (клональную экспансию) T-лимфоцитов. Зараженные вирусом или опухолевые клетки нередко не экспрессируют дополнительных стимулирующих молекул и поэтому не способны вызывать клональную экспансию эффекторных клеток. Это лишний раз подчеркивает важность высокоспециализированной системы антигенпредставляющих клеток (в том числе
дендритных) в запуске T-клеточного ответа.
Взаимодействие дендритных клеток с B- и T-лимфоцитами
Главные эффекторные клетки антиген-специфического иммунного ответа — B- и T-лимфоциты. Функция этих клеток зависит от дендритных клеток. Последние захватывают антигены, перерабатывают и переносят их на свою поверхность. Там, в комплексе с дополнительными стимулирующими молекулами,
они активируют T-лимфоциты. B-лимфоциты могут распознавать антиген после связывания с антигенраспознающими рецепторами B-лимфоцитов. Распознавание антигена T-лимфоцитами возможно только
после предварительной переработки и представления пептидных фрагментов антигена дендритными
клетками. T-лимфоциты могут связываться с пептидными фрагментами антигена через различные антигенраспознающие рецепторы, при этом комплекс пептидный антиген —HLA класса I активирует
T-лимфоциты CD8, а комплекс пептидный антиген —HLA класса II — T-лимфоциты CD4. Способность
дендритных клеток активировать T-лимфоциты зависит также от секреции стимулирующих цитокинов, в
том числе ИЛ-12 — ключевого цитокина в образовании и активации T-хелперов 1 типа и
NK-лимфоцитов.
Для стимуляции выраженного антиген-специфического T-клеточного ответа достаточно небольшого
числа дендритных клеток и антигена, что говорит о высокой иммуностимулирующей способности дендритных клеток. Экспрессия молекул адгезии и лектинов (в частности DC-SIGN) способствует агрегации
дендритных клеток и T-лимфоцитов и усиливает взаимодействие с антиген-связывающими рецепторами
T-лимфоцитов.
DC-SIGN — это лектин типа C, который связывается с лентивирусами (в том числе с вирусом иммунодефицита обезьян, ВИЧ-1 и ВИЧ-2), посредством gp120 и углеводов (49). Иммуногистохимические исследования показали, что DC-SIGN экспрессируется на дендритных клетках подслизистого слоя и кожи.
Полагают, что DC-SIGN играет роль в заражении ВИЧ через слизистые и при вертикальной передаче
инфекции. Было показано, что экспрессия DC-SIGN способствует проникновению ВИЧ в T-лимфоциты и
позволяет использовать корецепторы, если их экспрессия ограничена. Таким образом, DC-SIGN может
оказаться тем средством, с помощью которого ВИЧ проникает в дендритные клетки слизистых оболочек.
Зараженные дендритные клетки мигрируют в лимфоидную ткань, где ВИЧ передается лимфоцитам CD4.
Уже на ранней стадии инфекции репликация вируса в лимфоидной ткани очень активна (50, 51). В первой фазе ВИЧ-инфекции происходит всплеск виремии, за которым следует относительное снижение
концентрации вируса в крови. В это время в большом количестве появляются специфичные к ВИЧ цитотоксические T-лимфоциты, что совпадает с первым снижением концентрации вируса крови. В лимфоидной ткани вирусы захватываются сетью фолликулярных дендритных клеток. Главная мишень вируса —
макрофаги и активированные и покоящиеся лимфоциты CD4. На всем протяжении ВИЧ-инфекции лимфоидная ткань служит основным местом репликации ВИЧ. В лимфоидной ткани доля клеток, содержащих провирусную ДНК, в 5-10 раз выше, чем среди циркулирующих мононуклеаров крови, а репликация
ВИЧ в лимфоидной ткани на 1-2 порядка выше, чем в крови. Таким образом, основным резервуаром
ВИЧ служат лимфатические узлы.
После проникновения ВИЧ в покоящийся лимфоцит CD4 и завершения обратной транскрипции, вирусный геном представлен провирусной невстроенной ДНК. Для встраивания провирусной ДНК в геном
клетки-хозяина, и следовательно, для образования новых вирусов необходима активация T-лимфоцитов.
Поэтому лимфоидная ткань служит самой благоприятной средой для репликации ВИЧ. Тесный контакт
лимфоцитов CD4 и антигенпредставляющих клеток, наличие вирусов на поверхности фолликулярных
дендритных клеток и изобилие провоспалительных цитокинов (в частности, ИЛ-1, ИЛ-6 и ФНО?) способствуют началу репликации ВИЧ в инфицированных клетках и поддерживают ее в дальнейшем. Отметим, что ИЛ-1 и ФНО? индуцируют NF-kB, который связывается с длинным концевым повтором и запускает транскрипцию провируса. Важность антигензависимой активации лимфоцитов CD4 была подчеркнута несколькими исследованиями in vivo и in vitro, которые показали усиление репликации ВИЧ на
фоне вакцинации против столбняка или гриппа и при туберкулезе (52). Хотя польза вакцинации (например, профилактика гриппа или столбняка) у ВИЧ-инфицированных перевешивает риск временного повышения вирусной нагрузки, факт неоспорим: любая активация иммунной системы сопровождается усилением репликации ВИЧ.
На фоне ВААРТ в лимфоидной ткани значительно снижается число T-лимфоцитов, в которых идет активная репликация вируса (53). Однако, несмотря на успешное подавление виремии, во всех случаях остаются покоящиеся T-лимфоциты с латентной формой вируса (46). Эти клетки способны давать новый
виток вирусной репликации после отмены антивирусных препаратов.
При естественном течении ВИЧ-инфекции число лимфоцитов CD4 постепенно снижается, в то время как
концентрация ВИЧ в крови постепенно увеличивается. Серийные исследования лимфоидной ткани показали, что прогрессирование инфекции сопряжено с разрушением архитектоники лимфоидной ткани и
снижением захвата вирусных частиц. Различные иммуногистологические исследования показывают, что
паракортикальная зона лимфоузлов — это главная область, в которой начинается репликация ВИЧ (50,
51). Заражение соседних лимфоцитов CD4, а также их активация дендритными клетками определяют
распространение ВИЧ по лимфоидной ткани.
Как и при инфекции, вызванной вирусом иммунодефицита обезьян у резус-макак, при ВИЧ-инфекции на
всех ее стадиях репликация вируса и разрушение лимфоцитов CD4 происходят в собственной пластинке
слизистой и подслизистой кишечника активнее, чем в лимфоузлах (54, 55, 56). Это обусловлено тем, что
в кишечнике представлена основная популяция несущих рецептор CCR5 эффекторных клеток памяти
CD4, которые служат лучшей мишенью для репликации ВИЧ по сравнению со смешанной популяцией
лимфоцитов CD4 в лимфоузлах. Однако еще предстоит выяснить, играет ли выраженное снижение лимфоцитов CD4 в кишечнике важную роль в общем нарушении CD4-гомеостаза в течение заболевания.
Сегодня изучается также влияние ВИЧ-инфекции на тимус и его роль в снижении числа лимфоцитов
CD4. Последние работы показывают, что образование в тимусе лимфоцитов CD4 при ВИЧ-инфекции
снижается, особенно с возрастом, и что этот эффект связан с пролиферацией T-лимфоцитов в тимусе;
механизм этого остается неясным, так как тимоциты не экспрессируют CCR5 и не являются обязательной мишенью ВИЧ (57, 58).
Лимфоциты CD8 распознают «свой» антиген (пептидный фрагмент) в комплексе с HLA класса I антигенпредставляющей клетки, а лимфоциты CD4 — в комплексе с HLA класса II. Таким образом, специфический иммунный ответ на ВИЧ зависит от индивидуального набора антигенов HLA.
На антигенпредставляющих клетках связывание пептидов ВИЧ с углублением молекулы HLA класса I
может происходить по-разному. В результате активация лимфоцитов CD8 может быть достаточной, недостаточной или вообще не произойти. В крупных когортных исследованиях было проанализировано
влияние HLA на естественное течение ВИЧ-инфекции (быстрое прогрессирование и медленное прогрессирование инфекции). Связь между HLA и благоприятным течением инфекции прослеживалась примерно у 40% инфицированных из группы длительного непрогрессирующего течения инфекции. Протективным фактором от прогрессирования ВИЧ признана гомозиготность по HLA-Bw4. У гетерозигот по локусам HLA класса I иммунодефицит развивается медленнее, чем у гомозигот (59). Первое исследование
Kaslow в 1996 г. показало, что у носителей HLA-B14, B27, B51, B57 и C8 инфекция прогрессирует медленнее, а у носителей HLA-A23, B37 и B49 иммунодефицит развивается быстро (60). У всех ВИЧ-
инфицированных с HLA-B35 СПИД развивался не ранее, чем через 8 лет от заражения. Недавние исследования показали: у половых партнеров, несовместимых по HLA класса I, риск заражения ВИЧ при гетеросексуальных контактах ниже (61).
Исследования in vitro обнаружили у носителей HLA-B57 специфичные к вирусным пептидам лимфоциты, опосредующие ограниченную по HLA-B57 цитотоксичность. Однако вполне возможно, что протективный эффект некоторых аллелей HLA или пептидов ВИЧ, определяющих ограниченную по HLA цитотоксичность, не обязательно найдет применение в создании профилактических вакцин. Kaul и соавт. обнаружили, что лимфоциты CD8 контактировавших, но не заразившихся африканок и лимфоциты CD8
ВИЧ-инфицированных африканок распознавали разные эпитопы ВИЧ (62). Это говорит о том, что протективные эпитопы и иммуногенные могут не совпадать.
Антигены HLA класса II играют ключевую роль в специфическом к ВИЧ иммунном ответе, опосредованном лимфоцитами CD4. Rosenberg в 1997 г. впервые обнаружил у лицв с длительным непрогрессирующим течением инфекции специфичные к ВИЧ лимфоциты CD4, способные к пролиферации в ответ
на антигены ВИЧ (63). Протективное и неблагоприятное влияния различных аллелей HLA класса II изучены меньше, чем класса I. В когортных исследованиях заразившихся от матерей детей и ВИЧ-
инфицированных взрослых протективный эффект обнаружен у HLA-DR13 (64).
Рецепторы KIR (от англ. «killer cell immunoglobulin like receptors» — рецепторы типа иммуноглобулинов
клеток-киллеров) представляют лиганды, которые связываются с антигенами HLA класса I и путем активации или ингибирования рецепторов регулируют активацию NK-клеток. Показано, что полиморфизм по
генам KIR коррелирует с медленным или быстрым прогрессированием ВИЧ-инфекции, особенно, если в
анализ включали полиморфизм HLA класса I (65).
В целом, различный генетический полиморфизм оказывает влияние на течение ВИЧ-инфекции. Однако
сегодня нет оснований для того, чтобы рекомендовать генетические исследования у больных или лечение в зависимости от их результатов.
Цитотоксические T-лимфоциты (лимфоциты CD8) способны распознавать и элиминировать инфицированные вирусом клетки. Ряд исследований четко показывает, что цитотоксические T-лимфоциты играют
ключевую роль в сдерживании репликации ВИЧ и существенно влияют на прогрессирование болезни.
Однако значимая роль этих клеток в первичной защите от инфекции пока не доказана.
По сравнению с ВИЧ-инфицированными с быстрым падением числа лимфоцитов CD4 у больных с длительно непрогрессирующим течением болезни в изобилии обнаруживаются предшественники цитотоксических T-лимфоцитов, специфичные ко многим белкам ВИЧ. Связь более быстрого или медленного
прогрессирования инфекции с различными аллелями HLA может объясняться различной способностью
этих аллелей к представлению вирусных антигенов, от которой зависит сила иммунного ответа (см. выше).
Описаны случаи, когда через несколько лет стабильного течения ВИЧ-инфекции с выраженным
T-клеточным цитотоксическим ответом появлялись «ускользающие» от цитотоксических T-лимфоцитов
мутантные штаммы ВИЧ. Появление таких ускользающих мутантов совпадало с быстрым снижением
числа лимфоцитов CD4, что свидетельствует о защитной роли цитотоксических T-лимфоцитов (66).
Специфичные к ВИЧ цитотоксические T-лимфоциты были обнаружены у контактировавших с ВИЧ, но
не заразившихся людей. У ВИЧ-отрицательных гетеросексуальных половых партнеров ВИЧ-
инфицированных были обнаружены цитотоксические T-лимфоциты, специфичные к белку Nef, а у
ВИЧ-отрицательных медицинских работников, подвергшихся уколу загрязненной иглой — специфичные
к белку Env (67). К сожалению, даже сильный цитотоксический ответ не защищает инфицированных от
суперинфекции другим, близким штаммом ВИЧ (68).
Появление цитотоксических T-лимфоцитов совпадает не только со спадом виремии в начальной стадии
ВИЧ-инфекции, но и с перерывами в антиретровирусной терапии (особенно если она была начата на
ранней стадии инфекции).
Однако до сих пор неясно, почему сильный T-клеточный цитотоксический ответ в большинстве случаев
со временем ослабевает. Возможно, дело объясняется появлением «ускользающих» мутантов: распознанные ранее эпитопы перестают быть иммунодоминантными.
Белок Nef может подавлять экспрессию антигенов HLA класса I и тем самым препятствовать распознаванию инфицированных клеток цитотоксическими T-лимфоцитами. Цитотоксические T-лимфоциты определяются у большинства ВИЧ-инфицированных, однако непонятно, почему их недостаточно для по-
давления вируса. Интересно, что цитотоксическим T-лимфоцитам ВИЧ-инфицированных присущи не-
достаток перфорина и незрелый фенотип по сравнению с эффекторными клетками, направленными про-
тив цитомегаловируса (69), хотя их способность секретировать хемокины и цитокины может быть нор-
мальной. Другое недавнее исследование показало, что цитотоксическая активность специфичных к ВИЧ
лимфоцитов CD8 зависит от их способности одновременно продуцировать интерферон ? и ФНО? (70).
Лимфоциты CD8 также могут заражаться ВИЧ (71), правда среди специфичных к ВИЧ T-лимфоцитов
CD8 зараженных обнаружено не было. Не ясно, могут ли лимфоциты CD8 на какое-то время экспресси-
ровать молекулы CD4 и какие корецепторы хемокинов участвуют в проникновении ВИЧ в эти клетки.
Пролиферация и активация лимфоцитов CD8 зависит от антиген-специфичных T-хелперов CD4.
Rosenberg и соавт. показали, что начало ВААРТ во время острой лихорадочной фазы приводит к сохра-
нению ВИЧ-специфичного ответа лимфоцитов CD4, который не заметен у больных на более поздних
стадиях инфекции (63). ВИЧ поражает в первую очередь активированные лимфоциты CD4, а поскольку
специфичные к ВИЧ лимфоциты входят в число первых клеток, активируемых в ходе ВИЧ-инфекции,
они страдают одними из первых (Douek и соавт., 72). Поэтому пока не ясно, является потеря ВИЧ-
специфичных цитотоксических лимфоцитов следствием их внутреннего дефекта или же вторичным ре-
зультатом потери специфичных T-хелперов CD4.
В последние годы было разработано множество лечебных вакцин, большинство из которых изучалось на
макаках-резус, инфицированных вирусом иммунодефицита обезьян. Цель применения этих вакцин —
вызвать специфичный цитотоксический иммунный ответ, который бы изменил естественное течение ин-
фекции. Недавно Lu и соавт. опубликовали обнадеживающие результаты испытания у макак-резус, ин-
фицированных вирусом иммунодефицита обезьян, вакцины с аутологичными дендритными клетками, на
которые воздействовал инактивированный вирус (73). По сравнению с контрольной группой у вакцини-
рованных обезьян отмечено существенное снижение вирусной нагрузки и развитие специфического гу-
морального и клеточного иммунного ответа. Сейчас проходит предварительное испытание вакцины у 18
ВИЧ-инфицированных со стабильной вирусной нагрузкой, не получавших АРВ препаратов. Этим боль-
ным ввели вакцину с аутологичными дендритными клетками, на которые воздействовал инактивирован-
ный аутологичный вирус. В последующие 112 дней медиана снижения вирусной нагрузки составила
80%, у 8 больных низкая вирусная нагрузка сохранялась более года. Параллельно выявлены
gag-специфичные лимфоциты CD8 и ВИЧ-специфичные лимфоциты CD4, продуцирующие интерферон ?
и ИЛ-2 (74). Вакцины с аутологичными дендритными клетками служат перспективным методом имму-
нотерапии, однако нуждаются в дальнейших контролируемых клинических испытаниях.
Помимо цитотоксического действия на инфицированные клетки, полученные от ВИЧ-инфицированных,
лимфоциты CD8 обладают выраженной гуморальной ингибиторной активностью в отношении ВИЧ: по-
казано, что они подавляют репликацию ВИЧ в аутологичных и аллогенных культурах клеток (75). Не-
смотря на многочисленные усилия объяснение этой активности (активность «CAF») пока не найдено.
Возможно, что хотя бы отчасти она связана с хемокинами, в частности с MIP-1?, MIP-1?, RANTES (26),
IL-16 (76), MDC (77) и дефенсинами (78).
В зависимости от секретируемых цитокинов лимфоциты CD4 делят на T-хелперы 1 и 2 типа. T-хелперы 1 типа вырабатывают в основном интерлейкин-2 (ИЛ-2) и интерферон-?. Эти цитокины поддерживают эффекторные функции иммунной системы (цитотоксических T-лимфоцитов, NK-лимфоцитов, макрофа- гов). T-хелперы 2 типа вырабатывают преимущественно ИЛ-4, ИЛ-10, ИЛ-5 и ИЛ-6. Эти цитокины акти- вируют гуморальный иммунный ответ. T-хелперы 1 типа играют важную роль в образовании цитотокси- ческих T-лимфоцитов, поэтому появление специфичных к ВИЧ T-хелперов 1 типа рассматривают как протективную иммунную реакцию. Исследования in vitro показали, что после стимуляции вирусными антигенами Env (gp120/gp160) и пептидами T-лимфоциты контактировавших с ВИЧ, но не заразившихся людей секретировали ИЛ-2, а контрольные T-лимфоциты не контактировавших — нет (79). Подобные наблюдения сделаны и у подвергшихся уколу инфицированной иглой медицинских работников и у ново- рожденных, родившихся от ВИЧ-инфицированных матерей. Хотя это может указывать на протективную роль T-хелперов 1 типа, следует учесть и другую возможность: подобный иммунный ответ мог быть вы- зван контактом с дефектными, не способными к развитию инфекции, вирусными частицами и поэтому не обязательно подразумевает защиту от способного к репликации вируса.
Роль гуморального иммунного ответа в течении ВИЧ-инфекции изучена меньше. В экспериментах с ви-
русом иммунодефицита обезьян инъекция различных антител (включая нейтрализующие), предотвраща-
ла проникновение вируса через слизистую (80). Это говорит о том, что первичная защита от инфекции в
основном зависит от широкого гуморального иммунного ответа. При уже развившейся инфекции, напро-
тив, снижение числа B-лимфоцитов моноклональными антителами, не влияло на концентрацию вирус-
ной РНК в крови обезьян (81).
Медленное прогрессирование иммунодефицита наблюдалось у больных с высокими титрами антител к
p24 (82), стабильностью титров нейтрализующих антител к первичным и аутологичным штаммам вируса
(83) и отсутствием антител к некоторым эпитопам gp120 (84).
У больных с длительным непрогрессирующим течением инфекции отмечена тенденция к широкой ак-
тивности нейтрализующих антител к ряду первичных штаммов и устойчивость титров нейтрализующих
антител против аутологичного штамма вируса. Пока не ясно, играют ли нейтрализующие антитела за-
щитную роль или просто отражают сохранность относительно интактной иммунной системы. К числу
людей с высоким риском ВИЧ-инфекции — контактировавших, но не заболевших — по определению
относят лиц, у которых не определяются антитела к ВИЧ. Это определение подразумевает, что систем-
ный гуморальный ответ может и не играть ведущей роли в защите от инфекции. У этих людей на слизи-
стых обнаружены секреторные IgA к белкам ВИЧ, которые обычными методами не выявляются (85). Та-
ким образом, защита от ВИЧ-инфекции может определяться местными секреторными IgA, а не циркули-
рующими IgG. По некоторым наблюдениям, определенные антитела к ВИЧ-1 могут облегчать проникно-
вение вируса в лимфоциты CD4.
Наличие нейтрализующих антител у ВИЧ-инфицированных подтверждено рядом исследований. Однако
появление этих антител, по-видимому, запаздывает: ко времени, когда появляются антитела, в плазме
появляются и новые — устойчивые к ним — штаммы вируса. Таким образом, гуморальный ответ напо-
минает стрельбу по движущейся мишени — вирус постоянно ускользает от антител. Возможно, даль-
нейшее изучение этой проблемы позволит создать новые методы воздействия на вирус.
Расширение знаний в области патофизиологии ВИЧ-инфекции позволяет совершенствовать антиретро-
вирусную терапию и открывать новые перспективы — например, применение цитокинов (ИЛ-2 и дру-
гих) и терапевтическую вакцинацию. Однако самой важной задачей, определяющей дальнейшее изуче-
ние иммунопатогенеза ВИЧ-инфекции, остается создание профилактической вакцины — жизненно на-
сущного средства для борьбы с эпидемией, особенно в странах Африки и Юго-Восточной Азии.
Источник: www.eurasiahealth.org